Fermionic Environments¶
Here we model a single fermion coupled to two electronic leads or reservoirs (e.g., this can describe a single quantum dot, a molecular transistor, etc). The system hamiltonian, \(H_{sys}\), and bath spectral density, \(J_D\), are
We will demonstrate how to describe the bath using two different expansions of the spectral density correlation function (Matsubara’s expansion and a Padé expansion), how to evolve the system in time, and how to calculate the steady state.
Since our fermion is coupled to two reservoirs, we will construct two baths – one for each reservoir or lead – and call them the left (\(L\)) and right (\(R\)) baths for convenience. Each bath will have a different chemical potential \(\mu\) which we will label \(\mu_L\) and \(\mu_R\).
First we will do this using the built-in implementations of the bath expansions,
LorentzianBath and
LorentzianPadeBath.
Afterwards, we will show how to calculate the bath expansion coefficients and to use those coefficients to construct your own bath description so that you can implement your own fermionic baths.
Our implementation of fermionic baths primarily follows the definitions used by Christian Schinabeck in his dissertation ( https://opus4.kobv.de/opus4-fau/files/10984/DissertationChristianSchinabeck.pdf ) and related publications.
A notebook containing a complete example similar to this one implemented in BoFiN can be found in example notebook 4b.
Describing the system and bath¶
First, let us construct the system Hamiltonian, \(H_{sys}\), and the initial
system state, rho0:
from qutip import basis, destroy
# The system Hamiltonian:
e1 = 1. # site energy
H_sys = e1 * destroy(2).dag() * destroy(2)
# Initial state of the system:
rho0 = basis(2,0) * basis(2,0).dag()
Now let us describe the bath properties:
# Shared bath properties:
gamma = 0.01 # coupling strength
W = 1.0 # cut-off
T = 0.025851991 # temperature
beta = 1. / T
# Chemical potentials for the two baths:
mu_L = 1.
mu_R = -1.
# System-bath coupling operator:
Q = destroy(2)
where \(\Gamma\) (gamma), \(W\) and \(T\) are the parameters of
an Lorentzian bath, \(\mu_L\) (mu_L) and \(\mu_R\) (mu_R) are
the chemical potentials of the left and right baths, and Q is the coupling
operator between the system and the baths.
We may the pass these parameters to either LorentzianBath or
LorentzianPadeBath to construct an expansion of the bath correlations:
from qutip.nonmarkov.heom import LorentzianBath
from qutip.nonmarkov.heom import LorentzianPadeBath
# Number of expansion terms to retain:
Nk = 2
# Matsubara expansion:
bath_L = LorentzianBath(Q, gamma, W, mu_L, T, Nk, tag="L")
bath_R = LorentzianBath(Q, gamma, W, mu_R, T, Nk, tag="R")
# Padé expansion:
bath_L = LorentzianPadeBath(Q, gamma, W, mu_L, T, Nk, tag="L")
bath_R = LorentzianPadeBath(Q, gamma, W, mu_R, T, Nk, tag="R")
Where Nk is the number of terms to retain within the expansion of the
bath.
Note that we haved labelled each bath with a tag (either “L” or “R”) so that we can identify the exponents from individual baths later when calculating the currents between the system and the bath.
System and bath dynamics¶
Now we are ready to construct a solver:
from qutip.nonmarkov.heom import HEOMSolver
from qutip import Options
max_depth = 5 # maximum hierarchy depth to retain
options = Options(nsteps=15_000)
baths = [bath_L, bath_R]
solver = HEOMSolver(H_sys, baths, max_depth=max_depth, options=options)
and to calculate the system evolution as a function of time:
tlist = [0, 10, 20] # times to evaluate the system state at
result = solver.run(rho0, tlist)
As in the bosonic case, the max_depth parameter determines how many levels
of the hierarchy to retain.
As in the bosonic case, we can specify e_ops in order to retrieve the
expectation values of operators at each given time. See
System and bath dynamics for a fuller description of
the returned result object.
Below we run the solver again, but use e_ops to store the expectation
values of the population of the system states:
# Define the operators that measure the populations of the two
# system states:
P11p = basis(2,0) * basis(2,0).dag()
P22p = basis(2,1) * basis(2,1).dag()
# Run the solver:
tlist = np.linspace(0, 500, 101)
result = solver.run(rho0, tlist, e_ops={"11": P11p, "22": P22p})
# Plot the results:
fig, axes = plt.subplots(1, 1, sharex=True, figsize=(8,8))
axes.plot(result.times, result.expect["11"], 'b', linewidth=2, label="P11")
axes.plot(result.times, result.expect["22"], 'r', linewidth=2, label="P22")
axes.set_xlabel(r't', fontsize=28)
axes.legend(loc=0, fontsize=12)
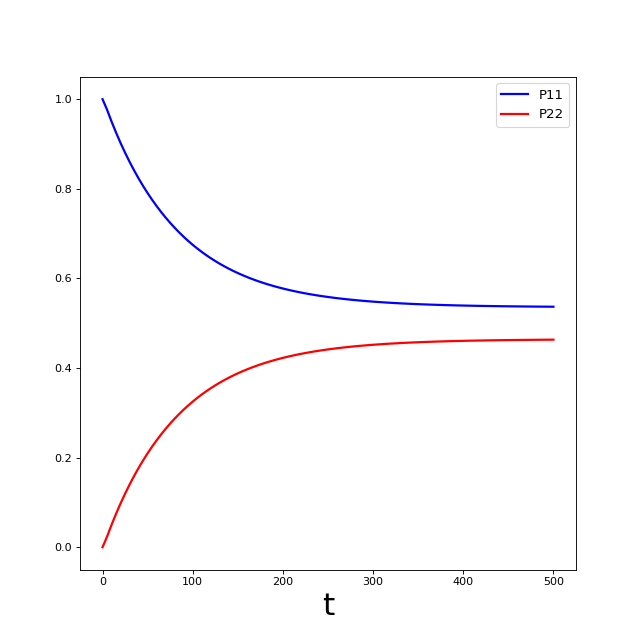
The plot above is not very exciting. What we would really like to see in this case are the currents between the system and the two baths. We will plot these in the next section using the auxiliary density operators (ADOs) returned by the solver.
Determining currents¶
The currents between the system and a fermionic bath may be calculated from the first level auxiliary density operators (ADOs) associated with the exponents of that bath.
The contribution to the current into a given bath from each exponent in that bath is:
where the \(\pm\) sign is the sign of the exponent (see the
description later in Padé expansion coefficients) and
\(Q^\pm\) is \(Q\) for + exponents and \(Q^{\dagger}\) for
- exponents.
The first-level exponents for the left bath are retrieved by calling
.filter(tags=["L"]) on ado_state which is an instance of
HierarchyADOsState and also provides access to
the methods of HierarchyADOs which describes the
structure of the hierarchy for a given problem.
Here the tag “L” matches the tag passed when constructing bath_L earlier
in this example.
Similarly, we may calculate the current to the right bath from the exponents tagged with “R”.
def exp_current(aux, exp):
""" Calculate the current for a single exponent. """
sign = 1 if exp.type == exp.types["+"] else -1
op = exp.Q if exp.type == exp.types["+"] else exp.Q.dag()
return 1j * sign * (op * aux).tr()
def heom_current(tag, ado_state):
""" Calculate the current between the system and the given bath. """
level_1_ados = [
(ado_state.extract(label), ado_state.exps(label)[0])
for label in ado_state.filter(tags=[tag])
]
return np.real(sum(exp_current(aux, exp) for aux, exp in level_1_ados))
heom_left_current = lambda t, ado_state: heom_current("L", ado_state)
heom_right_current = lambda t, ado_state: heom_current("R", ado_state)
Once we have defined functions for retrieving the currents for the
baths, we can pass them to e_ops and plot the results:
# Run the solver (returning ADO states):
tlist = np.linspace(0, 100, 201)
result = solver.run(rho0, tlist, e_ops={
"left_currents": heom_left_current,
"right_currents": heom_right_current,
})
# Plot the results:
fig, axes = plt.subplots(1, 1, sharex=True, figsize=(8,8))
axes.plot(
result.times, result.expect["left_currents"], 'b',
linewidth=2, label=r"Bath L",
)
axes.plot(
result.times, result.expect["right_currents"], 'r',
linewidth=2, label="Bath R",
)
axes.set_xlabel(r't', fontsize=28)
axes.set_ylabel(r'Current', fontsize=20)
axes.set_title(r'System to Bath Currents', fontsize=20)
axes.legend(loc=0, fontsize=12)
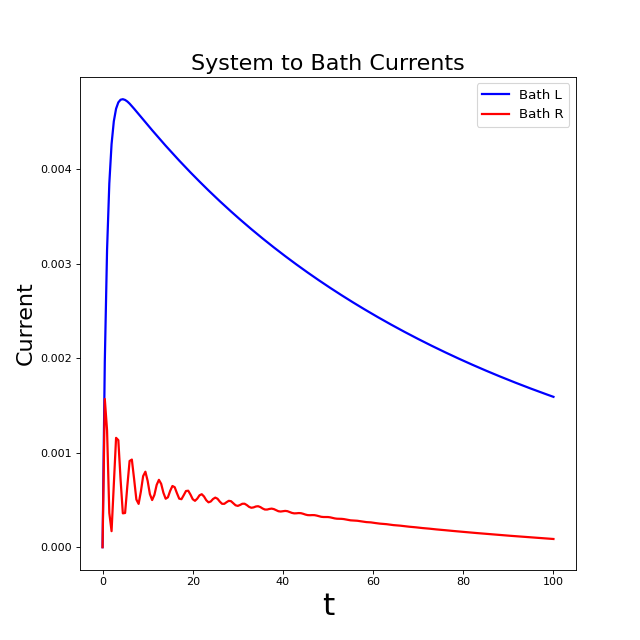
And now we have a more interesting plot that shows the currents to the left and right baths decaying towards their steady states!
In the next section, we will calculate the steady state currents directly.
Steady state currents¶
Using the same solver, we can also determine the steady state of the combined system and bath using:
steady_state, steady_ados = solver.steady_state()
and calculate the steady state currents to the two baths from steady_ados
using the same heom_current function we defined previously:
steady_state_current_left = heom_current("L", steady_ados)
steady_state_current_right = heom_current("R", steady_ados)
Now we can add the steady state currents to the previous plot:
# Plot the results and steady state currents:
fig, axes = plt.subplots(1, 1, sharex=True, figsize=(8,8))
axes.plot(
result.times, result.expect["left_currents"], 'b',
linewidth=2, label=r"Bath L",
)
axes.plot(
result.times, [steady_state_current_left] * len(result.times), 'b:',
linewidth=2, label=r"Bath L (steady state)",
)
axes.plot(
result.times, result.expect["right_currents"], 'r',
linewidth=2, label="Bath R",
)
axes.plot(
result.times, [steady_state_current_right] * len(result.times), 'r:',
linewidth=2, label=r"Bath R (steady state)",
)
axes.set_xlabel(r't', fontsize=28)
axes.set_ylabel(r'Current', fontsize=20)
axes.set_title(r'System to Bath Currents (with steady states)', fontsize=20)
axes.legend(loc=0, fontsize=12)
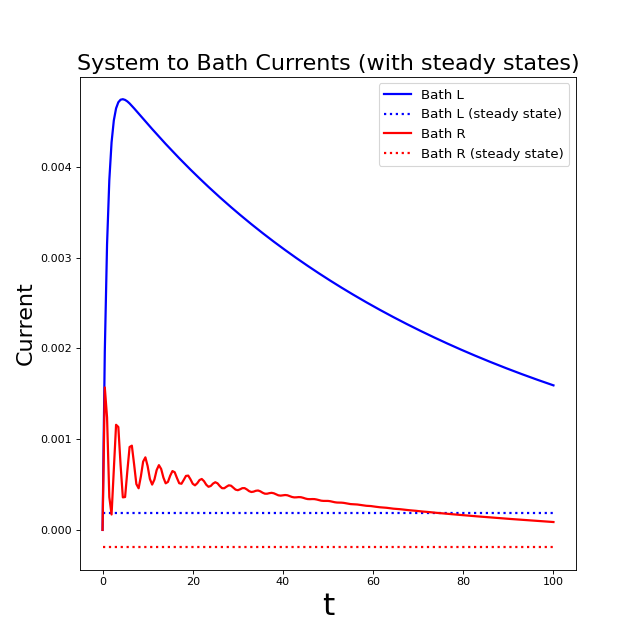
As you can see, there is still some way to go beyond t = 100 before the
steady state is reached!
Padé expansion coefficients¶
We now look at how to calculate the correlation expansion coefficients for the
Lorentzian spectral density ourselves. Once we have calculated the coefficients
we can construct a FermionicBath directly from
them. A similar procedure can be used to apply
HEOMSolver to any fermionic bath for which we can
calculate the expansion coefficients.
In the fermionic case we must descriminate between the order in which excitations are created within the bath, so we define two different correlation functions, \(C_{+}(t)\), and \(C_{-}(t)\):
where \(\sigma\) is either + or - and, \(f_F\) is the Fermi
distribution function, and \(J(\omega)\) is the Lorentzian spectral density
we defined at the start.
The Fermi distribution function is:
As in the bosonic case we can approximate this integral with a Matsubara or Padé expansion. For the Lorentzian bath the Padé expansion converges much more quickly, so we will calculate the Padé expansion coefficients here.
The Padé decomposition approximates the Fermi distribution as:
where \(k_l\) and \(\epsilon_l\) are coefficients defined in J. Chem Phys 133, “Efficient on the fly calculation of time correlation functions in computer simulations”, and \(Nk\) specifies the cut-off in the expansion.
Evaluating the integral for the correlation functions gives:
where:
and \(\beta = \frac{1}{T}\).
And now we calculate the same numbers in Python:
# Imports
from numpy.linalg import eigvalsh
# Convenience functions and parameters:
def deltafun(j, k):
""" Kronecker delta function. """
return 1.0 if j == k else 0.
def f_approx(x, Nk):
""" Padé approxmation to Fermi distribution. """
f = 0.5
for ll in range(1, Nk + 1):
# kappa and epsilon are calculated further down
f = f - 2 * kappa[ll] * x / (x**2 + epsilon[ll]**2)
return f
def kappa_epsilon(Nk):
""" Calculate kappa and epsilon coefficients. """
alpha = np.zeros((2 * Nk, 2 * Nk))
for j in range(2 * Nk):
for k in range(2 * Nk):
alpha[j][k] = (
(deltafun(j, k + 1) + deltafun(j, k - 1))
/ np.sqrt((2 * (j + 1) - 1) * (2 * (k + 1) - 1))
)
eps = [-2. / val for val in eigvalsh(alpha)[:Nk]]
alpha_p = np.zeros((2 * Nk - 1, 2 * Nk - 1))
for j in range(2 * Nk - 1):
for k in range(2 * Nk - 1):
alpha_p[j][k] = (
(deltafun(j, k + 1) + deltafun(j, k - 1))
/ np.sqrt((2 * (j + 1) + 1) * (2 * (k + 1) + 1))
)
chi = [-2. / val for val in eigvalsh(alpha_p)[:Nk - 1]]
eta_list = [
0.5 * Nk * (2 * (Nk + 1) - 1) * (
np.prod([chi[k]**2 - eps[j]**2 for k in range(Nk - 1)]) /
np.prod([
eps[k]**2 - eps[j]**2 + deltafun(j, k) for k in range(Nk)
])
)
for j in range(Nk)
]
kappa = [0] + eta_list
epsilon = [0] + eps
return kappa, epsilon
kappa, epsilon = kappa_epsilon(Nk)
# Phew, we made it to function that calculates the coefficients for the
# correlation function expansions:
def C(sigma, mu, Nk):
""" Calculate the expansion coefficients for C_\sigma. """
beta = 1. / T
ck = [0.5 * gamma * W * f_approx(1.0j * beta * W, Nk)]
vk = [W - sigma * 1.0j * mu]
for ll in range(1, Nk + 1):
ck.append(
-1.0j * (kappa[ll] / beta) * gamma * W**2
/ (-(epsilon[ll]**2 / beta**2) + W**2)
)
vk.append(epsilon[ll] / beta - sigma * 1.0j * mu)
return ck, vk
ck_plus_L, vk_plus_L = C(1.0, mu_L, Nk) # C_+, left bath
ck_minus_L, vk_minus_L = C(-1.0, mu_L, Nk) # C_-, left bath
ck_plus_R, vk_plus_R = C(1.0, mu_R, Nk) # C_+, right bath
ck_minus_R, vk_minus_R = C(-1.0, mu_R, Nk) # C_-, right bath
Finally we are ready to construct the
FermionicBath:
from qutip.nonmarkov.heom import FermionicBath
# Padé expansion:
bath_L = FermionicBath(Q, ck_plus_L, vk_plus_L, ck_minus_L, vk_minus_L)
bath_R = FermionicBath(Q, ck_plus_R, vk_plus_R, ck_minus_R, vk_minus_R)
And we’re done!
The FermionicBath can be used with the
HEOMSolver in exactly the same way as the baths
we constructed previously using the built-in Lorentzian bath expansions.